First trial steering Committee
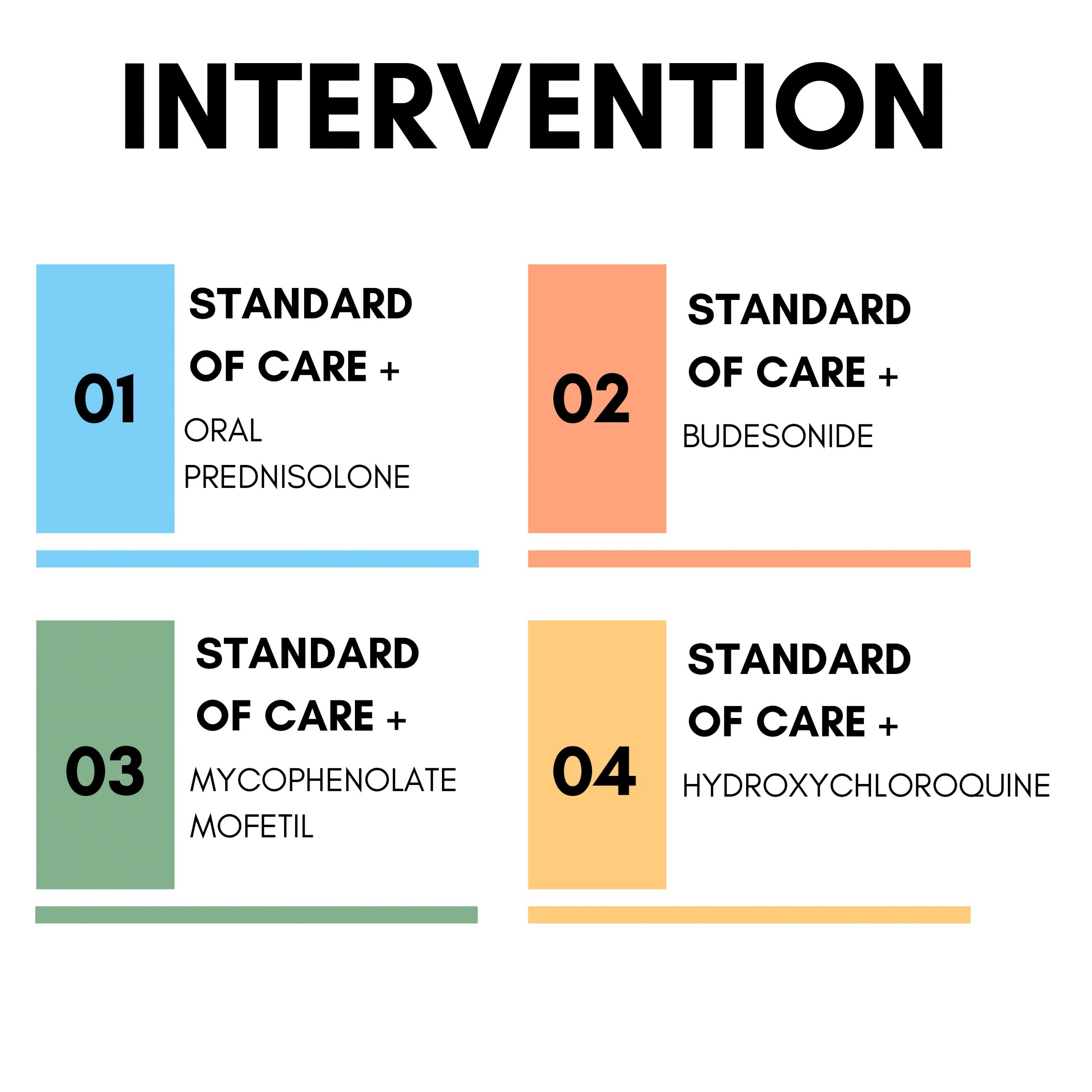
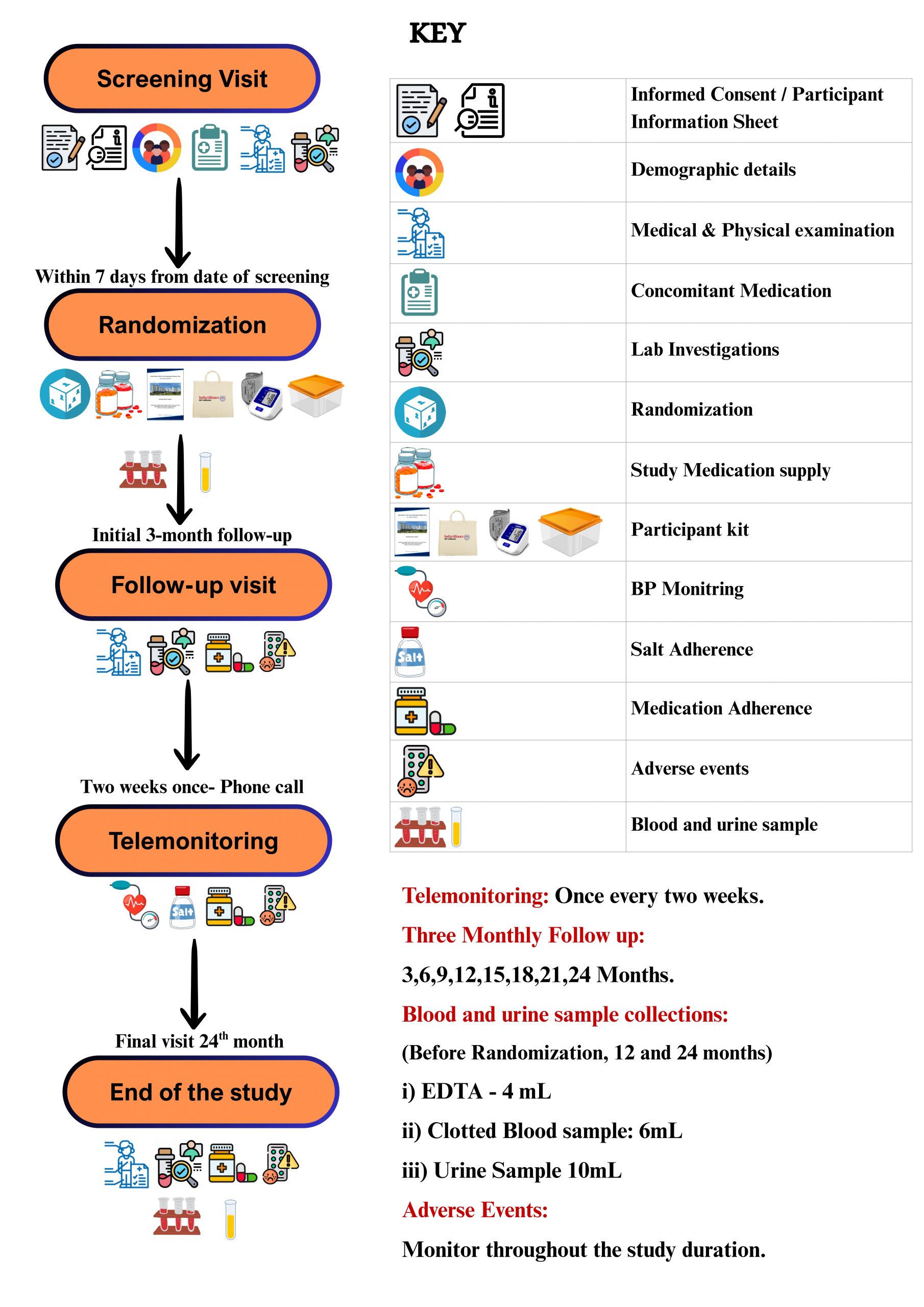
The First Trial Steering Committee Meeting was held as a event at the ISNCON 2023 venue, ITC Royal Bengal, on December 14, 2023. The primary focus of the meeting was Protocol Development and Consensus Building. PI’s from various sites participated, both in-person and virtually, to contribute their expertise and insights. Throughout the meeting, several key questions were discussed, encompassing various aspects of the trial protocol. These discussions were thorough and collaborative, ensuring that different perspectives were captured and considered. The outcomes of these discussions led to the implementation of several important protocol adjustments, which paves the way for current version of Master Protocol 1.6.